Here is an interesting exercice (and solution) we did today in Structural Bioinformatics class.
The goal is to fix an error in the following model:
Did you see this loop in the middle of the alpha-helix ?!
Before fixing it, these are Prosa plots of this model which confirm the error:
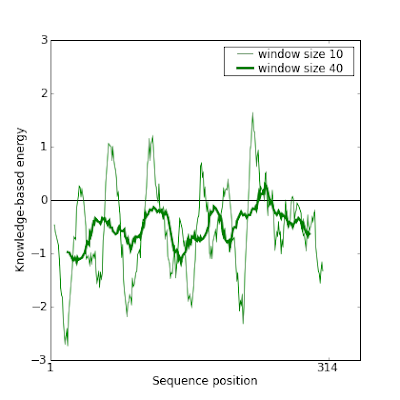
So here are the steps we followed to fix it. There are also interesting tricks to know to manipulate the data.
Getting the Amino Acids sequence
In order to get the corresponding AA sequence out of the PDB file, we simply splitted the PDB using our local tool which also generates the FASTA file out of PDB:
PDBtoSplitChain.pl -i wrong-model.pdb
Prepare the alignment for modeler
Then we produced an alignment between our target sequence against itself:
cat wrong-model.fa > homologs.fa
cat wrong-model.fa >> homologs.fa
Edit the homologs.fa file and name the first sequence "seq" and the second "model".
Create a clustal alignment with homologs.fa
clustalw homologs.fa
We have to locate the error (the loop of the alpha helix) in this alignment. For that purpose, we used Jmol and doing clicks on the AA of the loop we found the sequence Serine->Serine->Valine->Glutamic->Glutamic->Leucine->Leucine or ...SSVEELL...
We edit the alignment file and shift this subsequence to the right:
seq [...]QVAKSSVEELLLSQNSVKSL
model [...]QVAKSSVEELLLSQNSVKSL
Becomes:
seq [...]QVAKSSVEELLLSQNSVKSL-------
model [...]QVAK-------SSVEELLLSQNSVKSL
With this alignment we can produce a new model.
Generate a better model
Based on the alignment modified in previous section, we are going to produce a model and assess whether we improve it or not.
Convert the Clustal alignment file into a PIR file.
With the alignment in PIR format ready prepare the Modeler script:
from modeller import * # Load standard Modeller classes
from modeller.automodel import * # Load the automodel class
log.verbose() # request verbose output
env = environ() # create a new MODELLER environment to build this model in
# directories for input atom files
env.io.atom_files_directory = ['.', '../atom_files']
a = automodel(env,
alnfile = 'homologs.ali', # alignment filename
knowns = ('seq'), # codes of the templates
sequence = 'model') # code of the target
a.starting_model= 1 # index of the first model
a.ending_model = 3 # index of the last model
# (determines how many models to calculate)
a.make() # do the actual homology modeling
and create the model:
mod9v7 model-default.py
Assessment of the new model
This is the new model we just produced. Observed that the loop does not appear in the alpha helix:
And indeed this new model gives a much better result in Prosa:
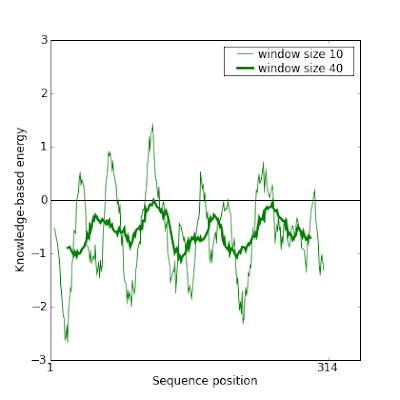
This manual modification of the alignment can be iteratively done to improve even more the model.








